一般我們拿到 10x 空間轉錄組數據分析的結果先看的肯定是 web_summary 網頁報告,因為從這個結果里面我們大概就能判斷你的數據好不好,不好的問題在哪里,數據到底能不能用等等。這里來詳細介紹一下怎么看 10x 空間轉錄組 web_summary 網頁版報告。
10x 空間轉錄組網頁版報告模板如下:
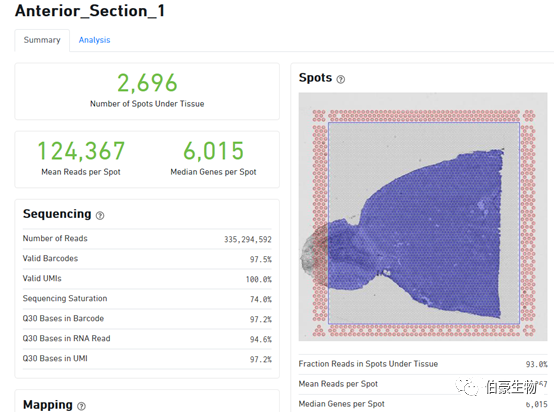
下面來詳細介紹一下每塊區域每個指標的含義。
Reads 總體情況統計區:

Number of Reads:樣本總的測序 reads 數,雙端測序這個是指一端的 reads 數,實際上算數據量需要用 reads*2* 讀長。
Valid Barcodes:barcode 校準后有效的 barcode 數占總的 reads 的比例,Space Ranger 會先嘗試糾正 barcode 序列中的序列錯誤,然后再進行統計。
Valid UMIs:有效的 UMI 數占總的 reads 的比例。
Sequencing Saturation: 測序飽和度值,就是在當前測序深度情況下,有多少比例的捕獲到的 mRNA 被測出來了,比如這這里的測序飽和度是 74%,說有 74% 的 mRNA 基因被檢測出來了,如果加大測序深度會有更多的 mRNA 被檢測出來。
Q30 Bases in Barcode:barcode 序列的 Q30 值
Q30 Bases in RNA Read:捕獲的 mRNA 序列的 Q30 值
Q30 Bases in UMI:UMI 序列的 Q30 值
Mapping 結果統計區:

Reads Mapped to Genome:比對到基因組上 reads 的比例
Reads Mapped Confidently to Genome:先進比對到基因組上 reads 的比例,也就是我們常說的 mapped uniquely reads,不過這里如果某條 reds 先進比對到一個基因的 exon 區,同時又比對到了一處非 exon 區,還是算先進比對到 exon 區的 reads。
Reads Mapped Confidently to Intergenic Regions:比對到先進基因間區的 reads 的比例
Reads Mapped Confidently to Intronic Regions:比對到先進內含子區的 reads 的比例
Reads Mapped Confidently to Exonic Regions:比對到先進外顯子區的 reads 的比例
Reads Mapped Confidently toTranscriptome: 比對到先進基因轉錄組上 reads 的比例,這一部分會包括剪切位點的 reads。這一部分的 reads 就是用來對 UMI 進行計數統計的。細心的朋友可能會發現這一部分的 reads 比例比 Reads Mapped Confidently to Exonic Regions 的值要低,這是因為有些基因的 exon 是有 overlap 的,處于 overlap 區域的 reads 是不進入 UMI 計數的。
Reads Mapped Antisense to Gene:比對到基因轉錄組的反義鏈區域的 reads 比例,這部分 reads 是沒有意義的。從這里我們也可以發現 10x 空間轉錄組建庫和比對有方向性的。
Spot 信息統計區:

Fraction Reads in Spots Under Tissue:比對到先進基因轉錄組上 reads(Reads Mapped Confidently to Transcriptome)有多少比例覆蓋在組織區域的 spot 上,這里是 93%,那就說明只有 7% 的 reads 分布在組織之外的灰色區域的。10x 軟件在這里有一個默認的閾值為 50%,認為這個比例值超過 50% 結果是正常的,低于 50% 則回到網頁 zuì 上面區域提示報錯信息(認為可能是透化不完全導致背景 RNA 過高或者是組織區域選的不合適)。從這個 50% 的閾值上我們也可以判斷 10x 的這個空間轉錄組技術還是存在一定缺陷的,它允許接近 50% 的 reads 散落在組織以外的區域,說明組織透化這一步想讓對應區域的 mRNA 完全都落入對應 spot 點里面去還是很難的。
Mean Reads per Spot:每個 spot 的平均 reads 數,10x 這里采用的是所以測序 reads 總是除以組織上檢測到的 spot 數(跟單細胞的統計方法是一樣的),理論上來說這樣統計是不合理的,因為總的 reads 包括沒有比對上的 reads、沒有 mapping 到轉錄本上的 reads、組織區域以外的 spot 上的 reads,所以是不能真實的反應每個 spot 上實際的 reads 數的。
Median Genes per Spot:每個 spot 的基因中位數
Total Genes Detected:檢測到的基因總數
Median UMI Counts per Spot:每個 spot 的中位 UMI 數
樣本信息區:

Sample ID:樣本 id
Chemistry:試劑版本
Slide Serial Number:Slide 信號和區域
Reference Path:參考基因組路徑
Transcriptome:基因組轉錄組版本
Pipeline Version:spaceranger 軟件版本
Analysis 區域
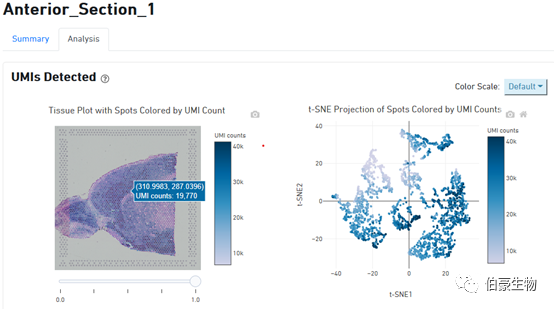
UMI 分布展示:左邊是圖像上 UMI 的分布,右邊是 tsne 降維可視化后的 UMI 的分布,鼠標放置到圖像上會現在對應的位置信息和對應 spot 上的 UMI count 數。從這個圖我們可以判斷 UMI 主要分布在組織的哪些區域,哪些區域沒有捕獲到 mRNA 或捕獲的 mRNA 特別少。
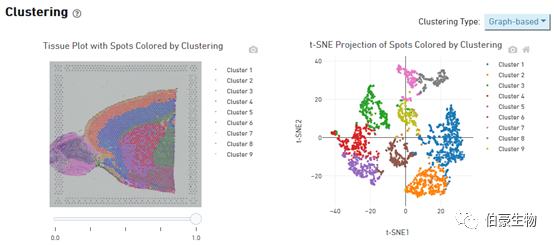
Cluster 的分布展示:左邊是 cluster 在組織圖像上的分布,右邊是 tsne 降維可視化后的 cluster 的分布,鼠標放置到圖像上會現在對應的位置信息和對應 spot 上的 cluster 值和該 cluster 占總的 spot 的比例。這個圖片上 cluster 分群在組織上的層次關系特別明顯。
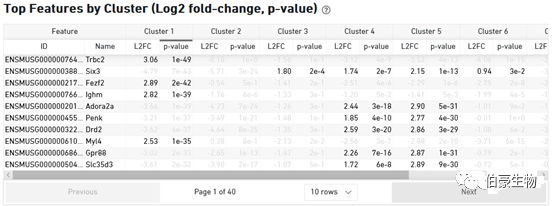
這一部分主要展示亞群的 top 基因的信息,因為不管是單細胞還是空間轉錄組基本上后面都會自己另外重新分析的,所以這部分和上面的 cluster 分布信息意義不大。
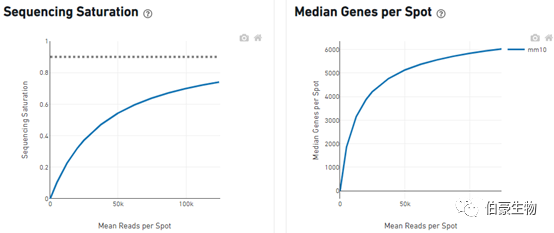
Sequencing Saturation(測序飽和度)
對 reads 進行隨機抽樣,觀察不同 spot 平均 reads 的情況下測序飽和度的分析,一直到實際的測序深度測序飽和度的值,理論上當所有轉化的 mRNA 轉錄本均已測序后,飽和度接近 1.0(100%),虛線表示測序到合理的飽和點位置,也就是說就是測序深度再高也不可能飽和度達到 100%。
Median Genes per Spot(sopt 點的中位基因)
也是對 reads 進行隨機抽樣,觀察不同 spot 平均 reads 的情況下 spot 的中位基因的值,曲線高點的斜率能反應增加測序深度能得到大的 spot 的中位基因數。
總結
對于 web_summary 的結果我們大概重點可以從下面幾個方面來看數據效果:
1、總的 spot 數,這個其實由組織的大小而定,沒有具體好壞的說法;
2、每個 spot 的中位基因數,中位基因數太少說明捕獲效果不好,有可能透化步驟條件不夠優化,當然也有可能是試劑或芯片的問題;
3、測序飽和度,每個點的 UMI 中位數,sopt 平均 reads 數,飽和度、sopt 平均 reads 數和中位 UMI 數都太低說明測序深度不夠,需要加大測序量。
4、基因組的比對率,比對率太低有可能是樣品污染;
5、組織 spot 上 reads 的比例,比對太低有可能透化時間不夠導致背景 RNA 過高,需要優化透化條件,也有可能組織區域選的不好,這個可以通過 LoupeBrowser 手動選擇組織區域。
更多伯豪生物人工服務:

