在發育過程中,細胞會對刺激做出反應,在整個生命過程中,從一種功能性“狀態”轉變為另一種功能性“狀態”。擬時序分析(pseudo-time),它指通過構建細胞間的變化軌跡來重塑細胞隨著時間的變化過程。用于擬時序分析的軟件和算法很多,目前文章中用到 多的是 Monocle 軟件,這也是是 CNS 主流期刊的常用軟件。
對于單細胞的數據做擬時序分析基本的流程是挑選一些感興趣并且可能有分化關系的亞群,然后來分析它們之間的分化關系。 那么對于空間轉錄組數據有沒有什么新花樣呢? 其實目前的空間轉錄組測序已發表的文章還沒太注重細胞分化這一內容,可能大家做空轉數據挖掘的時候還沒太把關注點放到分化關系這上面吧,那么今天就來給大家寫寫自己的突發奇想吧!
既然是做的空間轉錄組,那么我們就要有效的利用起來空間位置信息。選擇的亞群不能太離散。其次,我們可以分析相同細胞類型的亞群在空間位置的分化關系,也可以按照病理狀態分布的漸進變化來選擇區域做擬時序分化。
讀取 seurat 對象
library(monocle)
# 讀取前面保存的 seurat 對象文件
combin.data <- readRDS("combin.data.RDS")
這里我們選擇大腦皮層區的亞群作為示例進行細胞分化分析。
# 展示亞群分布
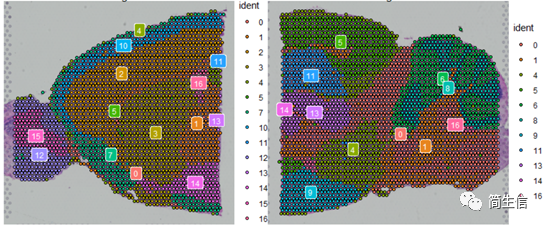
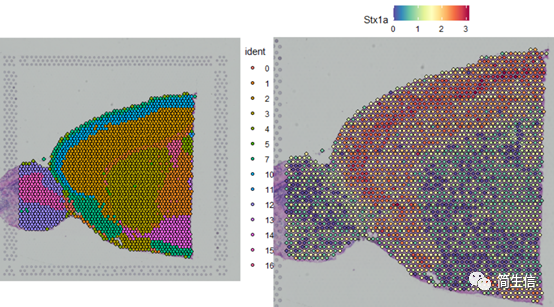
SpatialDimPlot(combin.data, label = TRUE, label.size = 3)
# 展示皮層 marker 的分布
SpatialFeaturePlot(combin.data, features = c("Stx1a"))
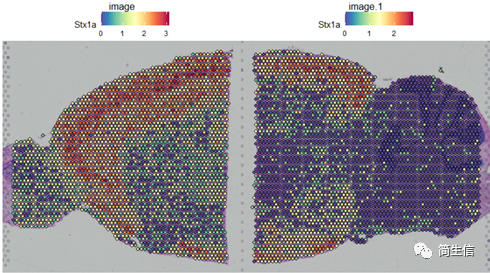
根據亞群和皮層 marker STX1A 的表達分布,我們選擇 2、5、7、9、10 號群作為皮層的亞群。
# 選擇要分析的亞群 subdata <- subset(combin.data, idents = c(2,5,7,9,10))
導入 seurat 對象,構建 CellDataSet 對象
這里我們使用 monocle2 做擬時序分析。monocle2 做擬時序分析需要三個矩陣文件:expression_matrix(表達矩陣,一般是 raw count)、phenoData(細胞 meta 文件,可以包括細胞的樣本、亞群等信息)、featureData(gene 的 meta 文件,注意要包括 gene_short_name 這一列)。
# 單細胞 count 文件、細胞類型注釋文件、基因注釋文件
expression_matrix = combin.data@assays$Spatial@counts
cell_metadata <- data.frame(group = combin.data[['orig.ident']],clusters = Idents(combin.data))
gene_annotation <- data.frame(gene_short_name = rownames(expression_matrix), stringsAsFactors = F)
rownames(gene_annotation) <- rownames(expression_matrix)
##### 新建 CellDataSet object
pd <- new("AnnotatedDataFrame", data = cell_metadata)
fd <- new("AnnotatedDataFrame", data = gene_annotation)
HSMM <- newCellDataSet(expression_matrix,
phenoData = pd,
featureData = fd,
expressionFamily=negbinomial.size())
monocle2 做擬時序分析主要包括三個步驟:
1、基因篩選。選擇要作為擬時序分化依據的基因,軟件官方提供了幾種可選的方法。
A、選擇不同時間點分化差異基因,這應該是比較理想的情況,需要你提供的數據是根據不同分化時間點取樣的;
B、選擇表達離散度高的基因,也就是在所有細胞里變化比較大的基因,如果只是想看某個亞群里細胞之間的分化選擇這種方法是比較合適的;
C、選擇亞群間差異大的基因,一般想比較多個亞群間的分化關系選擇這種方法效果會好一點;
D、選擇一些已知跟分化相關的基因。
2、數據降維。Monocle2 使用 DDRTree 的非線性重建算法對細胞進行排序。
3、細胞排序。根據細胞的降維后的結果給每個細胞計算一個分化時間。不過這里有一點需要注意,做細胞排序這一步可以自己指定分化起點,要不然算法會隨機選擇一端作為起點,也就是說計算出來的分化時間有可能是倒過來的,即起點是終點,終點是起點。
# 評估 SizeFactors
HSMM_myo <- estimateSizeFactors(HSMM)
# 計算離散度
HSMM_myo <- estimateDispersions(HSMM_myo)
# 計算差異
diff_test_res1 <- differentialGeneTest(HSMM_myo,fullModelFormulaStr = '~clusters', cores = 4)
# 選擇差異基因
ordering_genes <- subset(diff_test_res1, qval < 0.05)[,'gene_short_name']
# 基因過濾
HSMM_myo <- setOrderingFilter(HSMM_myo, ordering_genes)
# 降維
HSMM_myo <- reduceDimension(HSMM_myo, max_components=2, method = 'DDRTree')
# 細胞排序
HSMM_myo <- orderCells(HSMM_myo)
Monocle 結果可視化:
# 查看亞群分化關系
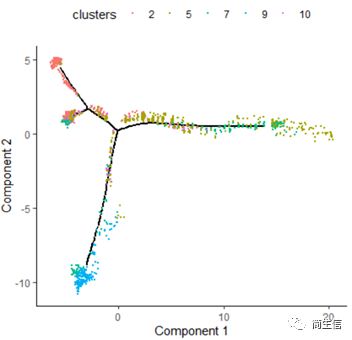
plot_cell_trajectory(HSMM_myo, color_by = "clusters",show_branch_points = F,cell_size =0.5)
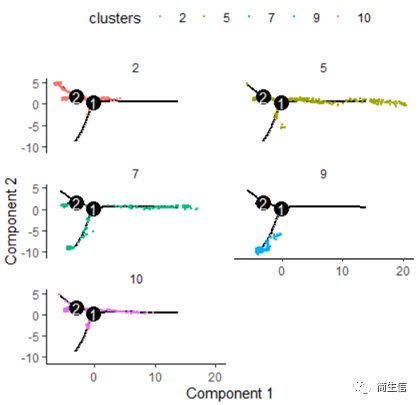
# 按亞群分開展示 plot_cell_trajectory(HSMM_myo, color_by = "clusters",cell_size =0.5) + facet_wrap(~clusters, nrow = 4)
# 分樣本展示 plot_cell_trajectory(HSMM_myo, color_by = "orig.ident",show_branch_points = F,cell_size =0.5)
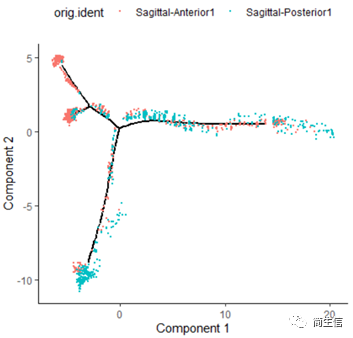
從亞群和樣本的分布來看其實還是體現了一定的樣本的異質性的,同一樣本的亞群更容易分布到同一個分支上,這里的 2、7、10 號群有點分不太開,這時候我們就要思考一下,對于空轉的數據有時候是不是單獨分析某個樣本上的分化是不是更合適一些。
選擇單個樣本的數據進行分析
這里我們選擇小鼠大腦 Sagittal-Anterior1 的樣本的皮層的亞群單獨進行分析。
#### 單獨繪制某個樣本的圖片
library("cowplot")
p1 <- SpatialPlot(combin.data,crop=FALSE,images='image')# 這里 Sagittal-Anterior1 樣本的鏡像圖片的名字是 image,具體名稱應跟前期讀取鏡像文件時設置的名字對應。
p2 <- SpatialFeaturePlot(combin.data, features = c("Stx1a"),images='image')
plot_grid(p1, p2)
選擇皮層 marker 高表達的 2、7、10 群進行擬時序分析。
# 選擇單個樣本 Sagittal-Anterior1 的亞群進行分析
subdata <- subset(combin.data, orig.ident == 'Sagittal-Anterior1')
subdata <- subset(subdata, idents = c(2,4,10))
# 單細胞 count 文件、細胞類型注釋文件、基因注釋文件
expression_matrix = subdata@assays$Spatial@counts
cell_metadata <- data.frame(group = subdata[['orig.ident']],clusters = Idents(subdata))
gene_annotation <- data.frame(gene_short_name = rownames(expression_matrix), stringsAsFactors = F)
rownames(gene_annotation) <- rownames(expression_matrix)
##### 新建 CellDataSet object
pd <- new("AnnotatedDataFrame", data = cell_metadata)
fd <- new("AnnotatedDataFrame", data = gene_annotation)
HSMM <- newCellDataSet(expression_matrix,
phenoData = pd,
featureData = fd,
expressionFamily=negbinomial.size())
# 評估 SizeFactors
HSMM_myo <- estimateSizeFactors(HSMM)
# 計算離散度
HSMM_myo <- estimateDispersions(HSMM_myo)
# 計算差異
diff_test_res1 <- differentialGeneTest(HSMM_myo,fullModelFormulaStr = '~clusters', cores = 4)
# 選擇差異基因
ordering_genes <- subset(diff_test_res1, qval < 0.05)[,'gene_short_name']
# 基因過濾
HSMM_myo <- setOrderingFilter(HSMM_myo, ordering_genes)
# 降維
HSMM_myo <- reduceDimension(HSMM_myo, max_components=2, method = 'DDRTree')
# 細胞排序
HSMM_myo <- orderCells(HSMM_myo)
結果可視化:
# 查看亞群分化關系
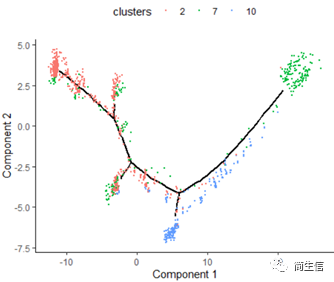
plot_cell_trajectory(HSMM_myo, color_by = "clusters",show_branch_points = F,cell_size =0.5)
# 按亞群分開展示
plot_cell_trajectory(HSMM_myo, color_by = "clusters",cell_size =0.5) + facet_wrap(~clusters, nrow = 4)
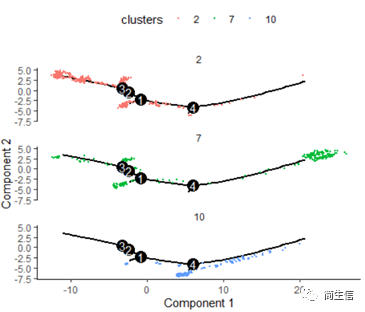
這里我們可以發現,之前選擇兩個樣本一起的 5 個亞群分析的時候這 3 個群是有點區分不開的,在這次重新分析的時候卻區分的很明顯。這也說明選擇的亞群范圍不一樣 monocle 得到的結果差異是很大的,究其原因可能是加入某些差異比較大的亞群或細胞后會進一步壓縮原本差異比較小的亞群之間的差異分布。因為是算法同時計算多組差異變化難免會出現這種情況,所以我們在一開始選擇亞群上就要稍微注意一點。
# 繪制分化時間
cell_Pseudotime <- data.frame(pData(HSMM_myo)$Pseudotime)
rownames(cell_Pseudotime) <- rownames(cell_metadata)
plot_cell_trajectory(HSMM_myo, color_by = "Pseudotime",cell_size =0.5)
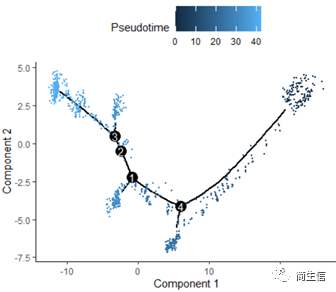
從分化時間的分布來看三個亞群的分化大概順序是從 7 號群一 >10 號群一 >2 號群,由于 monocle 的判斷的分化起點是隨機的,所以也有可能是倒過來的順序。
我們再來看一下皮層 marker 基因 Stx1a 的在分化過程中的表達分布
# 繪制 Stx1a 基因分化時間的變化
data_subset <- HSMM_myo['Stx1a',]
plot_genes_in_pseudotime(data_subset, color_by = "clusters")
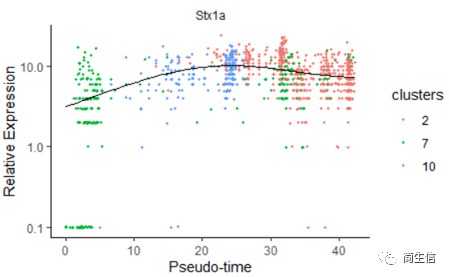
marker 基因的表達分布基本是由低到高再降低的趨勢。
下面重點來了!我們把細胞的分化時間對應到組織切片上。
# 把分化時間對應到到組織切片位置上
combin.data[['Pseudotime']] <- 0
combin.data[['Pseudotime']][rownames(cell_Pseudotime),] <- cell_Pseudotime
SpatialFeaturePlot(combin.data, features = c("Pseudotime"),images='image')
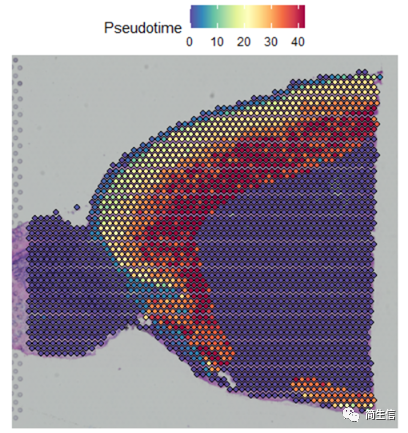
這時候我們就發現結果有點意思了,皮層細胞的分化在組織切片上是從外向內的方向的,當然也有可能實際上是反過來的,也就是從里往外的,總之就是細胞的分化是存在一定的空間位置規律的。
接著,我們再來看一下細胞分化過程中表達變化顯著的基因有哪些。
# 分析分化時間顯著變化的基因
diff_test_res2 <- differentialGeneTest(HSMM_myo[ordering_genes,],fullModelFormulaStr = "~sm.ns(Pseudotime)",cores = 4)
# 選擇 top50 基因繪圖
top50_gene = diff_test_res2[order(diff_test_res2$qval),][1:50,'gene_short_name']
plot_pseudotime_heatmap(HSMM_myo[top50_gene,], num_clusters = 6,cores = 1,show_rownames = T)
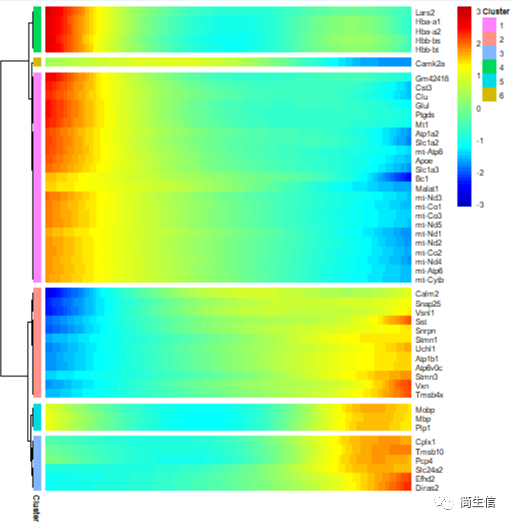

從 top50 差異基因的情況來看,血紅蛋白基因和線粒體基因在小鼠大腦皮層切片外層的表達比較高。我們也可以把所有差異基因都輸出來,將不同表達趨勢的基因模塊分別進行功能富集,這樣就可以知道隨著皮層細胞的在空間位置的分化哪些功能發生了變化。
sig_gene_names <- subset(diff_test_res2, qval < 0.05)[,'gene_short_name']
heatmap = plot_pseudotime_heatmap(HSMM_myo[sig_gene_names,], num_clusters = 6,cores = 1,show_rownames = F,return_heatmap=T)
###get cluster info
row_cluster = cutree(heatmap$tree_row,k=6)
write.table(row_cluster,file=paste("_gene_clusters.xls",sep=""),sep="\t",row.names=T)
細胞更有意思呢!因為示例數據里沒有病理信息,如果結合病理特征來做擬時序分化分析應該能發現更多有價值的東西!
轉載:簡生信(侵刪)

更多伯豪生物人工服務:

